
Druck-Version
Hintergrundwissen
Wie groß ist die genetische Varianz beim Menschen?
Was der Haplotyp über die menschliche Evolution verrät

Wie stark sich das menschliche Genom von dem beispielsweise der Schimpansen unterscheidet, kann man mittlerweile auch in der Bild-Zeitung nachlesen. Aber wie groß ist die Varianz des menschlichen Genoms? Das sogenannte HapMap-Projekt (aktueller Nachfolger: 1000-Genome-Projekt) sucht und dokumentiert Sequenzunterschiede in menschlichen Genomen.
Titelbild: Australopithecus africanus. José Braga; Didier Descouens, Mrs Ples, CC BY-SA 4.0.
{kind=link}
Der Großteil des Genoms ist bei allen Menschen identisch, aber durchschnittlich alle 1000 bis 1200 Basenpaare (bp) findet man einen Unterschied. Dies kann eine Deletion oder Insertion eines oder mehrerer Basen sein, aber meistens ist der Unterschied nur, welche Base an der Stelle steht. Zur Verdeutlichung: Eine Sequenz an einer Stelle des Genoms eines bestimmten Menschen könnte lauten:
...AGGTCAGT...
Wenn man die gleiche Stelle des Genoms bei verschiedenen anderen Menschen untersucht, kann es sein, dass man auch die folgenden Varianten dieses Abschnitts findet:
...AGGCCAGT...
...AGGGCAGT...
...AGGACAGT...
Die Auftreten von Sequenzunterschieden an einer bestimmten Stelle des Genoms bezeichnet man als Single Nucleotide Polymorphisms (SNPs; gesprochen: Snips) und jede einzelne "Schreibweise" stellt ein Allel dar. Da die Abschnitte zwischen den SNPs identisch sind, muss man, um die gesamte Genomsequenz eines Menschen zu kennen, theoretisch nur alle Stellen mit SNPs bestimmen.
...CAG....GTCA.....GTA...
...GAG....GCCA.....GTA...
...GAG....GGCA.....GTT...
...TAG....GACA.....GTT...
Würden in dem obigen Beispiel mit vier Allelen jeweils die Basen an den aufeinanderfolgenden SNPs (in rot) bestimmt, wüsste man die gesamte Sequenz der untersuchten (haploiden) Genome in diesem Bereich (da Menschen diploid sind, d. h. einen doppelten Chromosomensatz haben, von denen der eine vom Vater, der andere von der Mutter stammt, können sich die Sequenzen der beiden Chromosomensätze unterscheiden).
Die einzelnen SNPs kommen nicht in allen Kombinationen vor. Wenn in dem obigen Beispiel an der ersten Stelle C, G oder T stehen kann und an der zweiten T, C, G oder A, könnten theoretisch 12 verschieden Kombinationen auftreten. Tatsächlich findet man aber häufig nur bestimmte Kombinationen, in dem obigen Beispiel wäre das C an erster Stelle mit T an der zweiten, G nur mit C oder G, T nur mit A. Grund dafür ist, dass durch sexuelle Rekombination nahe beieinander liegende Bereich selten getrennt werden.
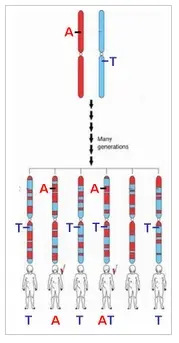
In der Abbildung ist zur Verdeutlichung ein Chromosomenpaar dargestellt, auf beiden ist jeweils ein Allel mit "A" bzw. "T" markiert. Ohne sexuelle Kombination würden die Allele auf diesen Chromosomen getrennt voneinander vererbt werden, kein Nachfahre würde beide Allele bekommen (es sei denn, eines der beiden Allel würde zufällig durch Mutation neu entstehen, was sehr unwahrscheinlich ist). Durch sexuelle Rekombination während der ersten Reifeteilung zur Produktion von Ei- bzw. Spermazellen (Crossing-over) können aber Teile beider Chromosomen ausgetauscht werden, so dass in Nachfolgegenerationen einzelne Menschen auch beide Allele auf einem Chromosomen tragen (im Beispiel: der mit "AT" markierte Mensch) oder keins von beiden.
In der Abbildung kann man auch erkennen, dass ein gewisser Abschnitt um das "T" herum bei allen Nachfahren vorhanden ist, die das "T"-Allel geerbt haben. SNPs, die in diesem Abschnitt liegen, sind bei einem Großteil der Menschen, die auch das "T" erhalten haben, gleich. Diese Abschnitte von SNPs, die verlinkt sind und in der Regel gemeinsam vererbt werden, bezeichnet man als Haplotyp und das "T" wäre ein sogenannter "Tag-SNP", da man nur schauen muss, ob ein Individuum ein "T" an dieser Stelle hat, um mit hoher Wahrscheinlichkeit auch die benachbarten SNPs zu kennen.
Das HapMap-Projekt versucht, eine Haplotypen-Bank aufzubauen und genau solche Tag-SNPs zu identifizieren. Dazu werden von insgesamt 270 Menschen aus verschiedenen Populationen (in Europa, China, Afrika, Japan) die SNPs bestimmt – im Oktober wurde die Phase II des Projekts veröffentlicht, mit über 3 Millionen SNPs (The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851-861. 2007). Basierend auf diesen Daten wird vermutet, dass es momentan insgesamt 9 – 10 Mio. relativ verbreitete SNPs gibt.
Grund für diese Datensammlung ist, dass man beispielsweise bei Menschen mit Multipler Sklerose (oder einer anderen Krankheit) mit Hilfe von Tag-SNPs relativ einfach die Haplotypen bestimmen und so feststellen könnte, ob bestimmte Haplotypen häufiger vorkommen als in der "Normal"-Bevölkerung. Gene, die von solchen "abweichenden" Haplotypen betroffen sind, könnten an der Entstehung der jeweiligen Krankheit beteiligt sein und dann genauer untersucht werden.
Aus den gesammelten Daten kann man auch viele Rückschlüsse auf den Verlauf und die Geschichte der menschlichen Evolution ziehen. Je länger eine Population besteht (je mehr Generationen), umso häufiger ist die Durchmischung von Chromosomen durch sexuelle Rekombination und umso größer die Wahrscheinlichkeit, dass auch nahe zusammenliegende Bereiche/SNPs von einander getrennt werden. Das Resultat sind viele verschiedene, kürzere Haplotypen. Von den untersuchten Populationen weist die afrikanische die meisten Haplotypen auf, die im Durchschnitt auch kürzer sind als die in den anderen Populationen.
Dies ist ein weiterer Beleg für die allgemein anerkannte "Out-of-Africa"-Hypothese, nach der der Ursprung der Menschheit in Afrika liegt. Die ausgewanderten Gruppen, aus denen die anderen Populationen hervorgegangen sind, haben nur einen Bruchteil der zu dem Zeitpunkt vorhandenen Variation "mitgenommen" und es ist weniger Zeit für die Entstehung neuer Varianten gewesen, so dass alle anderen Populationen längere und nicht so viele verschiedene Haplotypen aufweisen.
Als Folge dessen ist es in Europa, China oder Japan wahrscheinlicher, für bestimmte Abschnitte des Genoms homozygot zu sein, also von beiden Eltern den gleichen Haplotyp zu erben und folglich auf beiden Chromosomen eines Paars identische Allele aufzuweisen.
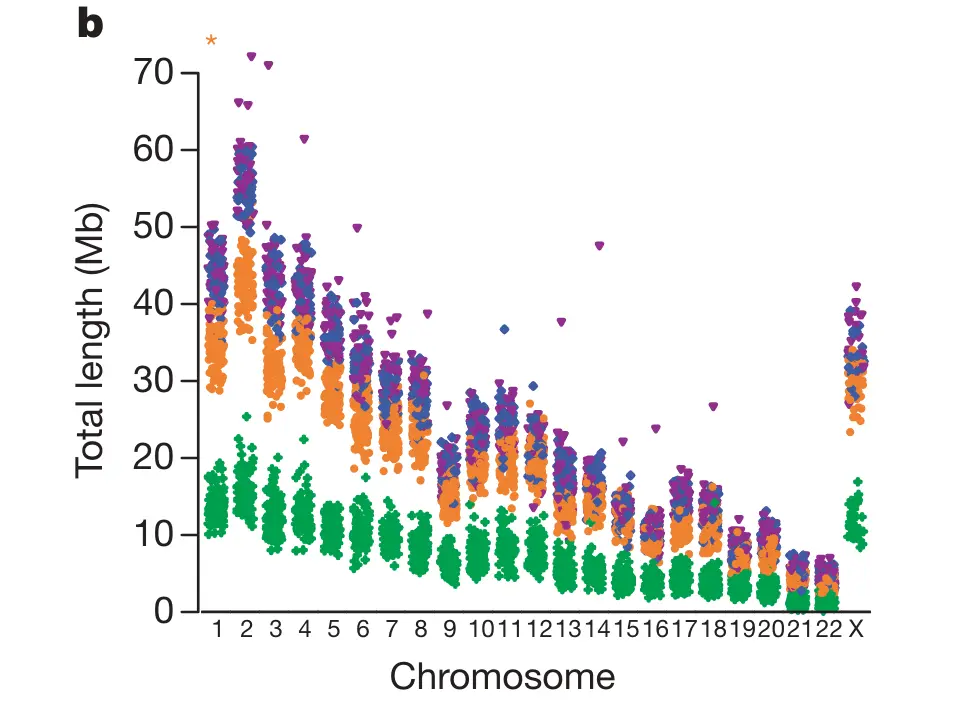
In dem oben zitierten Nature-Artikel von 2007 findet sich auch die nebenstehende Darstellung dieses Sachverhalts. Für die Abbildung haben sie bestimmt, wie groß die homozygoten Bereiche des Genoms bei jedem der untersuchten Individuen sind (für die einzelnen Chromosomen getrennt aufgetragen; das X-Chromosom konnte natürlich nur bei Frauen auf homozygote Abschnitte untersucht werden). Grün: Afrika, orange: Europa, violett: China, blau: Japan. Die "Gefahr", unwissentlich einen "Verwandten" zu heiraten, ist also für Japaner und Chinesen viel größer als für Afrikaner...
Anlass dieses Beitrags war eigentlich ein neuer Artikel über Daten aus dem HapMap-Projekt [BARREIRO et al. (2008) Natural selection has driven population differentiation in modern humans. Nature Genetics doi:10.1038/ng.78], der sehr interessant ist, aber das werde ich angesichts der Länge, die dieser Beitrag schon hat, besser in einem neuen Artikel machen.
Um die anfängliche Frage noch zusammenfassend zu beantworten: Jeder Mensch hat mit jedem anderen nicht-verwandten Menschen mindestens 99,9 % der Genomsequenz gemeinsam, die restlichen 0,1 % sorgen für unterschiedliches Aussehen, erhöhen oder senken das Risiko, an bestimmten Krankheiten zu erkranken oder besser/schlechter auf manche Medikamente anzusprechen, befähigen die einen zum "Zungerollen", während andere das nicht können und sorgen insgesamt dafür, dass das Leben etwas interessanter wird.
Literatur
The International HapMap Consortium (2008) A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851-861.
Autorin: Sabine Schu


