![]()
PDF-Version
Neues aus der Forschung
Chemische Evolution: Wie die Natur die Vorliebe für linkshändige Aminosäuren und rechtshändige Zucker entdeckte
Neue Erkenntnisse zur Lösung des Chiralitäts-Problems
Die Natur hat eine manische Vorliebe für Aminosäuren und Zucker einer ganz bestimmten Konfiguration: Nahezu alle Kettenmoleküle des Lebens, wie Proteine, Nukleinsäuren und Kohlenhydrate, enthalten nur eine von zwei möglichen Molekülsorten, die sich wie Bild und Spiegelbild verhalten, sich aber nicht durch Drehen zur Deckung bringen lassen. Konventionell werden solche "Spiegelzwillinge" mit Bezeichnungen wie links- und rechtshändig voneinander unterschieden. So kommen in der belebten Welt fast ausschließlich "linkshändige" Aminosäure- und "rechtshändige" Zuckermoleküle vor. Der Grund dafür ist weitgehend rätselhaft, denn chemisch müssen auf der frühen Erde beide Molekülsorten in gleichen Mengen entstanden sein und gleich gut miteinander reagiert haben.
Was sorgte für die dauerhafte Diskriminierung eines Spiegelzwillings? In der Fachzeitschrift "Nature" berichten Forscher von der Entdeckung gleich mehrerer Mechanismen, die eine plausible Erklärung liefern könnten.
Einführung: Was sind links- und rechtshändige Moleküle?
Hat jedes Molekül einen Spiegelzwilling? Nein, natürlich nicht. Ein Wassermolekül beispielsweise lässt sich problemlos so drehen, dass es mit seinem eigenen Spiegelbild deckungsgleich ist. Folglich ergibt hier der Begriff der Händigkeit (im Fachjargon Chiralität genannt) keinen Sinn. Bei anorganischen Verbindungen ist das die Regel, doch in der organischen Chemie (Kohlenstoff-Chemie) sieht dies oft anders aus. Das liegt an den vier Valenzen des Kohlenstoffatoms und an der Stabilität der Kohlenstoff-Kohlenstoff-Bindung, was eine enorme strukturelle Vielfalt zulässt.
Bild und Spiegelbild von organischen Molekülen lassen sich voneinander unterscheiden, wenn das Molekül mindestens ein Kohlenstoff-Atom mit vier unterschiedlichen Bindungspartnern (Substituenten) enthält (Abb. 1). Solche Kohlenstoffatome werden stereochemische Zentren oder Chiralitätszentren genannt. Der Begriff chiral stammt aus dem Griechischen und bedeutet so viel wie händig.1) Denn auch unsere beiden Hände verhalten sich wie Bild und Spiegelbild zueinander. Analog dazu wird von links- und rechtshändigen Molekül-Varianten gesprochen. Der Fachausdruck für spiegelbildlich aufgebaute Moleküle lautet Enantiomere.
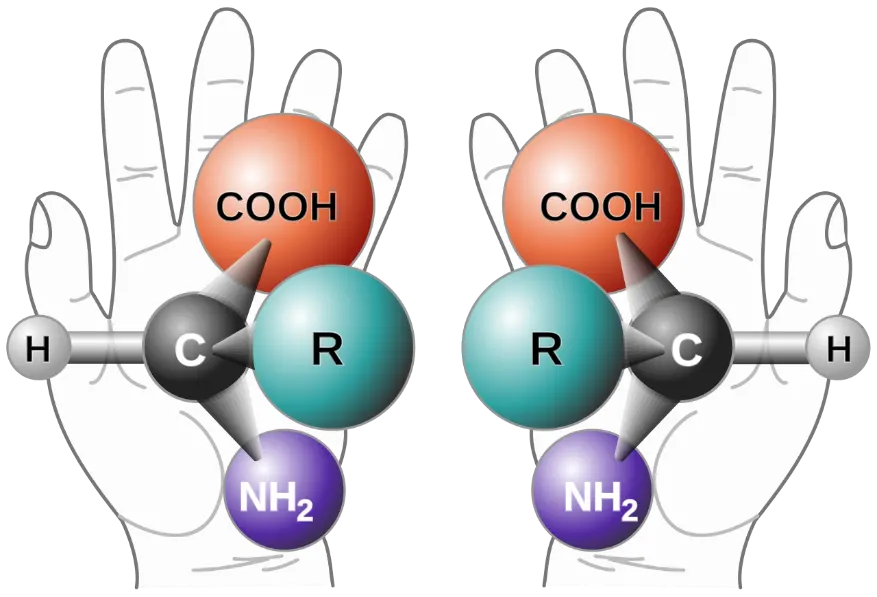
Abb. 1 Die beiden Enantiomere (sich wie Bild und Spiegelbild zueinander verhaltenden Varianten) eines Aminosäure-Moleküls unterscheiden sich im räumlichen Aufbau, ähnlich wie rechte und linke Hand. Sie werden daher als chiral (händig) bezeichnet. Konventionell unterscheidet man linkshändige von rechtshändigen Molekülen. Weiteres dazu im Text. Bildquelle: Wikipedia, als gemeinfrei gekennzeichnet.
Chemisch verhalten sich Enantiomere gleich, in einigen physikalische Eigenschaften, wie etwa der optischen Aktivität, sind sie jedoch verschieden. So dreht das eine Enantiomer linear polarisiertes Licht (genauer gesagt, dessen Polarisationsebene) im Uhrzeigersinn nach rechts, das andere nach links. Man spricht dann bei ersterem von der (+)-Form der chemischen Verbindung. Die andere Sorte wird folgerichtig als die (-)-Form bezeichnet.
Bekannter und wichtiger als die Benennung der Drehrichtung ist allerdings eine Nomenklatur, die auf den Chemiker Emil FISCHER zurückgeht. Um die komplizierte dreidimensionale Struktur des Moleküls anschaulich darzustellen, ersann FISCHER im Jahr 1891 Projektionsregeln, die eine zweidimensionale Darstellung erlauben. Aus ihr geht klar hervor, ob wir es mit der rechtshändigen oder linkshändigen Form zu tun haben (s. Abb. 2 und 3). Handelt es sich um das rechtshändige Molekül, wird dem Verbindungsnamen ein "D" vorangestellt (das "D" stammt vom lateinischen Begriff dexter für rechts). Die linkshändige Form nennt sich L-Form (L von laevulus, lat. für links). Die Kürzel "L" und "D" sind also stereochemische Deskriptoren, die anzeigen, welche Händigkeit das betreffende Molekül hat.
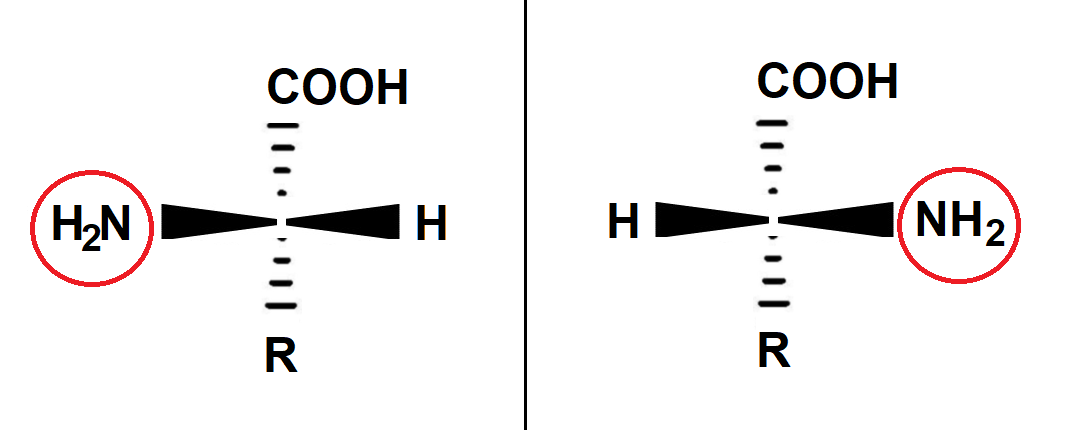
Abb. 2 Die beiden Enantiomere eines Aminosäure-Moleküls in der Keilstrich-Formel. Nach FISCHER wird das Molekül so dargestellt, dass die längste Kohlenstoffkette in der Vertikalen verläuft und das am höchsten oxidierte C-Atom (hier die COOH-Gruppe) oben liegt. Die schwarzen Keile deuten an, dass die waagerecht liegenden Substituenten (die Aminogruppe NH2 und das H-Atom) aus der Ebene herausragen. Die senkrecht stehende Carboxylgruppe (COOH) und der chemische Rest (R) liegen hinter der Betrachter-Ebene, was die gestrichelten Keile andeuten. Im Kreuzungspunkt befindet sich das stereochemische Zentrum – ein Kohlenstoff-Atom mit vier verschiedenen Substituenten. Liegt die Aminogruppe in der Projektion auf der linken Seite, handelt es sich um die L-Form. Liegt sie rechts, liegt die D-Form vor. In der FISCHER-Projektion lässt man die Keile meist weg und zeichnet sich kreuzende Linien (vgl. Abb. 3).
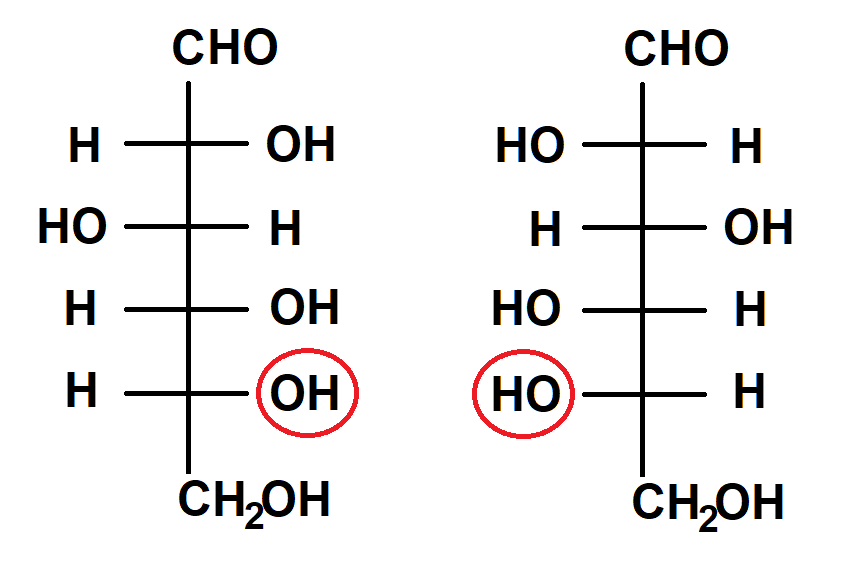
Abb. 3 D-Glucose (links) und L-Glucose (rechts) in der FISCHER-Projektion. Hier existieren mehrere, nämlich gleich vier stereochemische Zentren! In solchen Fällen bestimmt per Konvention die Lage der OH-Gruppe des am weitesten von der Aldehydgruppe (CHO) entfernten Chiralitätszentrums (hier rot eingekreist) den stereochemischen Deskriptor.
Gelegentlich wird hinter dem stereochemischen Deskriptor auch die Drehrichtung der Ebene des polarisierten Lichtes vermerkt, z. B. "D(+)-Glucose". Die Drehrichtung hat aber keine praktische Bedeutung mehr, sodass man sie meist weglässt. Aus ihr lässt sich auch nicht die Konfiguration der Enantiomere ermitteln. Letztere ist eine inhärente Struktureigenschaft der Moleküle, Drehrichtung und Drehwert hängen dagegen auch von äußeren Faktoren wie der Temperatur, dem Lösungsmittel usw. ab. Die Deskriptoren "D" und "L" dürfen also nicht mit der Drehrichtung verwechselt werden.
Obwohl FISCHERs D,L-Nomenklatur veraltet und durch die komplexere R-S-Konvention von CAHN, INGOLD und PRELOG ersetzt worden ist, ist sie in der Biochemie noch immer üblich. Das liegt sicher an ihrer Einfachheit bzw. daran, dass die zweidimensionale Projektion komplexer Moleküle sehr anschaulich ist.
Die Vorliebe der Natur für L-Aminosäuren und D-Zucker
Wie eingangs betont, erzeugen Lebewesen keine Enantiomeren-Gemische (razemische Gemische), sondern jeweils nur ein Enantiomer: Sämtliche Proteine (lange Kettenmoleküle aus Aminosäuren) sind aus linkshändigen Aminosäuren (L-Aminosäuren) aufgebaut. Bei den Nukleinsäuren greift die Natur ausschließlich auf rechtshändige Zucker (D-Zucker) zurück. Dagegen erhält man immer Gemische aus links- und rechtshändigen Molekülen, wenn man Aminosäuren und Zucker klassisch im Labor herstellt. Eine Trennung der beiden Enantiomere ist aufwendig und erfordert chemisches Knowhow.
Umso akuter stellt sich die Frage: Welche Faktoren waren für die Symmetrie-Brechung verantwortlich, die im Verlauf der Entstehung des Lebens zur Verwendung von nur einer Enantiomeren-Sorte führte?
Die Ungleichverteilung von D- und L-Bausteinen
Die Fachwelt ist sich darin einig, dass der bis heute propagierten Vorliebe der Natur für eine Sorte Biomoleküle anfangs eine Ungleichverteilung der D- und L-Bausteine in der "Ursuppe" vorausgegangen sein muss. Es muss also in den wie auch immer gearteten Reaktionsräumen ein Überschuss an einem bestimmten Enantiomer vorgelegen haben, der die Produktion von Kettenmolekülen begünstigte, die entweder nur aus D- oder L-Bausteinen zusammengesetzt waren. Solche Makromoleküle bezeichnet man als homochiral. Wie entstand diese Ungleichverteilung?
Eine mögliche Antwort darauf liefern Meteoriten, die einen Überschuss der L-Enantiomere von Aminosäuren enthalten (CRONIN & PIZZARELLO 1997). Darüber hinaus enthalten einige Meteoriten Zuckerderivate mit einem großen Überschuss der D-Enantiomere (COOPER & RIOS 2016). Möglicherweise erzeugen (kosmische) Strahlung, Magnetfelder oder thermische Schockereignisse ein Übergewicht der "gewünschten" Enantiomere. Welche Faktoren es auch sein mögen, die Befunde sind starke Indizien dafür, dass abiotische Wege existieren, um Aminosäuren und Zucker auf nichtrazemische Weise zu erzeugen (BURTON & BERGER 2018).
Möglicherweise haben meteoritische Verbindungen auch direkt das Enantiomeren-Profil der späteren Biomoleküle beeinflusst und zu deren Homochiralität beigetragen. Übrigens zeigen auch Mineralien oft einen Kristallhabitus mit chiralen Oberflächen (etwa Calcit) oder Symmetrien (Quarz), sodass sie bevorzugt bestimmte Enantiomere adsorbieren können (HAZEN 2006; LEE et al. 2022).
Nun neigen chirale Verbindungen allerdings zur Razemisierung, das heißt der anfängliche Enantiomeren-Überschuss (in der Literatur mit "ee" abgekürzt, von enantiomeric excess) geht im Lauf der Zeit allmählich wieder verloren. Auch wenn dieser Prozess normalerweise sehr langsam abläuft, braucht es einen Mechanismus, der die Ungleichverteilung verstärkt und weiter vorantreibt.
Propagation durch autokatalytische Prozesse
Eine mögliche Lösung des Problems sind sogenannte autokatalytische Prozesse, die in den letzten Jahren zusehends die Aufmerksamkeit der Forscher auf sich ziehen. Darunter versteht man chemische Reaktionen, bei denen das Produkt (hier: ein bestimmtes Enantiomer) seine eigene Herstellung unterstützt (katalysiert).
Eine der bemerkenswertesten autokatalytischen Reaktionen diesbezüglich ist die sogenannte Soai-Reaktion, einer Alkylierungsreaktion von Pyrimidin-5-Carboxaldehyd. Der in dieser Reaktion entstehende Alkohol ist chiral, wobei jedes der Enantiomere die eigene Bildung katalysiert. Ausgehend von einem winzigen Enantiomeren-Überschuss erzeugt der Pyrimidylalkohol als asymmetrischer Autokatalysator eine nahezu enantiomerenreine Lösung (>99,5 % ee) in hohen Ausbeuten (SOAI et al. 1995).
Der organische Autokatalysator der Soai-Reaktion lässt sich übrigens durch präbiotisch relevantere Katalysatoren ersetzen, darunter die Mineralien Quarz, Gips, Zinnober und Natriumborat – auch sie erzeugen enantiomerenreine Lösungen in hohen Ausbeuten (MATSUMOTO et al. 2017).
Homochirale Proteine durch Thiol-Katalyse
Was die Bildung von Proteinen durch enantioselektive Verknüpfung von Aminosäuren anbelangt, so brachten jüngst zwei Publikationen Licht ins Dunkel. Die erste stammt aus der Arbeitsgruppe des Chemikers Matthew POWNER (SINGH et al 2022).
Die Forscher entdeckten, dass eine Reihe von Schwefelalkoholen (Thiolen), deren Existenz präbiotisch plausibel ist, die Verknüpfung einzelner Aminosäuren mit Aminosäure-Vorläufern (sogenannten Aminonitrilen) katalysieren (s. Abb. 4). Diese Reaktion ist ein bemerkenswertes und seltenes Beispiel für Organokatalyse in Wasser. Da sie zudem mit allen in Organismen vorkommenden Aminosäuren gleichermaßen gut funktioniert, bieten sie einen plausiblen Weg zur Entstehung der ersten Proteine.
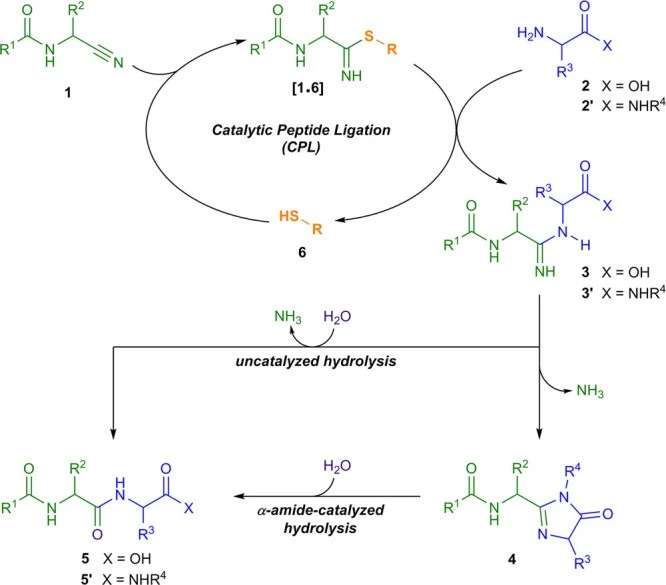
Abb. 4 Reaktionsschema der Thiol-katalysierten Peptid-Verknüpfung (CLP) nach SINGH et al. (2022). Mehrere Eigenschaften machen diese Reaktion als Mechanismus für die präbiotische Peptidsynthese äußerst attraktiv: Sie verwendet einfache präbiotische Reaktanden,2) ist selektiv für α-Amidonitrile (und damit für proteinogene Peptide), erzeugt hohe Peptidausbeuten unter Bedingungen, unter denen Peptide sehr stabil sind, und ist orthogonal zur (biologischen) Phosphataktivierung. Zudem läuft die Kettenbildung im Wasser ab. Bildquelle: SINGH et al. (2022).
Das Team von POWNER überprüfte nicht, ob seine schwefelbasierten Katalysatoren enantioselektiv waren. Diese Aufgabe übernahm die Forschungsgruppe der Chemikern Donna BLACKMOND (DENG et al. 2024). Deren Resultate waren zunächst ernüchternd, denn unter dem Einfluss der Thiol-Katalysatoren entstanden bevorzugt heterochirale Mischprodukte. "Paradoxerweise", so BLACKMOND und Kollegen, stelle "diese heterochirale Selektivität jedoch einen Mechanismus für die Enantiomeren-Anreicherung sowohl der homochiralen Dipeptide als auch der übrigen Substrate dar".
Setzt man nämlich ungleiche Anteile der Enantiomere (etwa 60% L- und 40% D-Aminosäuren) ein, verbleibt ein Übergewicht an nicht umgesetzten L-Aminosäuren in der Lösung. Durch einen physikalischen Effekt wird der Prozess der Enantiomeren-Anreicherung noch verstärkt: Das Team fand heraus, dass die heterochiralen Dipeptide wesentlich rascher aus der Lösung ausfallen als die homochiralen Dipeptide.
Als nächstes hoffen die Forscher herauszufinden, ob dieselben Mechanismen größere Peptide und Proteine in Richtung Linkshändigkeit verzerren – und ob sie auch für die rechtshändigen Zucker in der RNA gelten. Wenn ja, könnten die neuen Mechanismen erklären, wie das Leben selbst die eine spiegelbildliche Form annahm und nicht die andere. Die bisherigen Ergebnisse scheinen sehr vielversprechend.
Literatur
BURTON, A.S. & BERGER, E.L. (2018) Insights into abiotically-generated amino acid enantiomeric excesses found in meteorites. Life 8, 14.
COOPER, G. & RIOS, A.C. (2016) Enantiomer excesses of rare and common sugar derivatives in carbonaceous meteorites. Proc. Natl. Acad. Sci. USA 113, E3322–E3331.
CRONIN, J.R. & PIZZARELLO, S. (1997) Enantiomeric excesses in meteoritic amino acids. Science 275, S. 951–955.
DENG, M.; YU, J. & BLACKMOND, D. (2024) Symmetry breaking and chiral amplification in prebiotic ligation reactions. Nature 626, S. 1019–1024.
FODEN, C.; ISLAM, S.; GARCIA, C.A.F.; MAUGERI, L.; SHEPPARD, T. & POWNER, M. (2020) Prebiotic synthesis of cysteine peptides that catalyze peptide ligation in neutral water. Science 370, S. 865-869.
HAZEN, R.M. (2006) Mineral surfaces and the prebiotic selection and organization of biomolecules. Am. Mineral. 91, S. 1715–1729.
LEE, C.; WEBER, J.M.; RODRIGUEZ, L.E.; SHEPPARD, R.Y.; BARGE, L.M.; BERGER, E.L. & BURTON, A.S. (2022) Chirality in organic and mineral systems: a review of reactivity and alteration processes relevant to prebiotic chemistry and life detection missions. Symmetry 14, 460.
MATSUMOTO, A.; KAIMORI, Y.; UCHIDA, M.; OMORI, H.; KAWASAKI, T.; SOAI, K. (2017) Achiral inorganic gypsum acts as an origin of chirality through its enantiotopic surface in conjunction with asymmetric autocatalysis. Angew. Chem. Int. Ed. 56, S. 545–548.
SINGH, J.; WHITAKER, D.; THOMA, B.; ISLAM, S.; FODEN, C.S.; ALIEV, A.E.; SHEPPARD, T.D. & POWNER, M.W. (2022) Prebiotic catalytic peptide ligation yields proteinogenic peptides by intramolecular amide catalyzed hydrolysis facilitating regioselective lysine ligation in neutral water. J Am Chem Soc. 144, S. 10151–10155.
SOAI, K.; SHIBATA, T.; MORIOKA, H. & CHOJI, K. (1995) Asymmetric autocatalysis and amplification of enantiomeric excess of a chiral molecule. Nature 378, S. 767–768.
Fußnoten
[1] Daneben gibt es noch weitere Formen der Chiralität, wie beispielsweise die helikale Chiralität einer rechts- oder linksdrehenden Schraube. Solche Formen brauchen uns hier nicht zu interessieren.
[2] Für die Fachleute unter den Lesern: Die regioselektive N-Acetylierung des α-Aminonitrils, die zur Ausgangsverbindung (1) in Abb. 4 führt, ist der entscheidende initiale Schritt bei der abiotischen Peptidsynthese (FODEN et al. 2020). Abiotisch erfolgt sie leicht mithilfe von Thioessigsäure in Gegenwart von Hexacyanoferrat-(III). Die Acetylgruppe verhindert unerwünschte Nebenreaktionen und den durch Hydantoin, Diketopiperazin und Imidazol ausgelösten Peptidabbau.
Autor: Martin Neukamm


